What does our group do?
What does our group do?
Atmospheric aerosols are solid or liquid particles suspended in atmospheric air. We are studying the formation and properties of the smallest of the aerosol particles.

Selected facts about aerosols
- Aerosol particles are found in all parts of the Earth's atmosphere, both in polluted urban areas, and also naturally in remote areas, e.g. as a consequence of volcanic and biological activity
- The size of aerosol particles ranges from small molecular clusters of less than 1 nanometer up to rain drops of more than 1 millimeter
- Aerosols can harm human health and reduce visibility
- Aerosol particles can both warm and cool the climate, their combined net effect is cooling
- Every cloud droplet has an aerosol particle as a seed, and especially the effect of aerosols via cloud formation is perhaps the largest uncertainty in predicting climate change
- Up to 50-90% of the total particle number, and 30-50% of the cloud condensation nuclei, are produced by clustering from gas-phase molecules
Members of the computational aerosol physics group are working on deepening the theoretical understanding of the formation of the very smallest molecular clusters in the atmosphere. For this, we use a palette of different modeling approaches from inexpensive hard sphere collision theories and bulk liquid approximations to high level quantum mechanical treatment of each electron. Currently, our most active areas of research are
- Computational Chemistry Based Thermodynamics
- Cluster Population Simulations
- Chemistry of Clusters
- Molecular Dynamics Simulations
- Classical Nucleation Theory
which are all briefly described in the following. We have projects suited for B.Sc. or M.Sc. theses, but also welcome new ideas. If You are interested in our research and would like to work with us, please feel free to contact Prof. Hanna Vehkamäki, or any other member of our group.
Computational Chemistry Based Thermodynamics
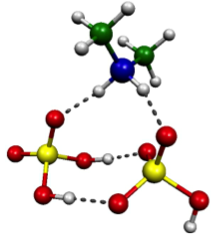
Experimental measurements and computational methods based on classical physics are not alone able to accurately describe the first steps of aerosol formation. On the sub-nanometer scale where the initial clustering steps occur, there is no substitute for quantum mechanics, especially for the atmospheric systems where clustering involves proton transfer reactions. We use methods based on calculating the electronic structure of systems (with various approximations) to explore the structures and formation energetics of small molecular clusters. These computational chemistry methods yield thermodynamic data which can be used to assess cluster evaporation rates. This data may be used directly in our model of cluster population dynamics, or may be parametrized for use combined with macroscopic classical nucleation theory. Currently we are working on extending the molecular basis of the simulated clusters, amongst others by including oxidised organic compounds and water in our simulations.
Related recent publications from the group:
- Elm et al., Physical Chemistry Chemical Physics, Vol 19, pp 4877-4886, 2017
- Elm et al., Journal of Physical Chemistry A, Vol 120, pp 3693-3700, 2016
- Elm et al., Journal of Physical Chemistry A, Vol 120, pp 2240-2249, 2016
- Elm et al., Journal of Physical Chemistry A, Vol 119, pp 8414-8421, 2015
- Ortega et al., Atmospheric Chemistry and Physics, Vol 14, pp 7995-8007, 2014
- Henschel et al., Journal of Physical Chemistry A, Vol 118, pp 2599-2611, 2014
- Leverentz et al., Journal of Physical Chemistry A, Vol 117, pp 3819–3825, 2013
- Ryding et al., Journal of Physical Chemistry A, Vol 116, pp 4902–4908, 2012
- Ortega et al., Atmospheric Chemistry and Physics, Vol 12, pp 225-235, 2012
Cluster Population Simulations
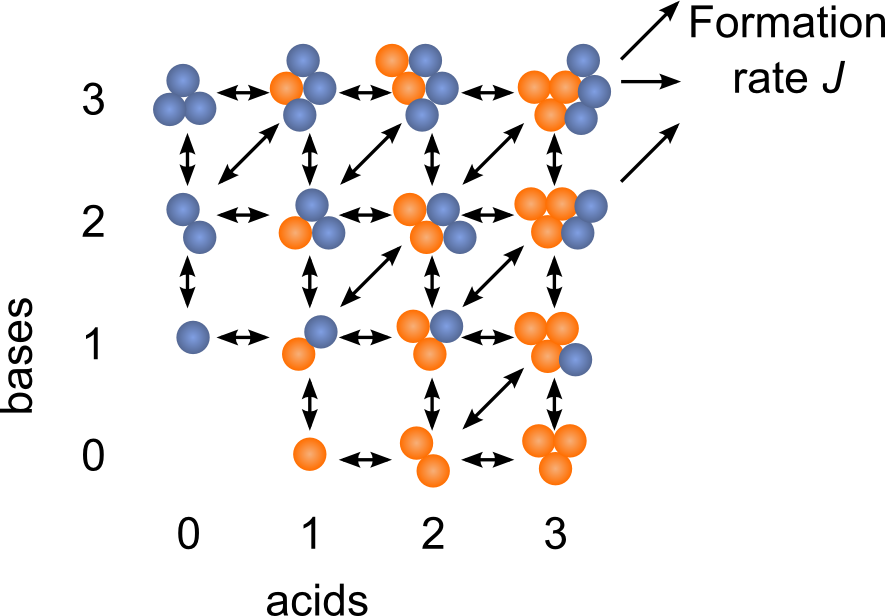
The Atmospheric Cluster Dynamics Code (ACDC) is a cluster population dynamics model developed in our group. ACDC generates the birth-death equations governing the formation and loss processes of the clusters, and solves them numerically to find the time evolution of all cluster concentrations. The kinetic processes considered in the equations include all possible collisions and evaporations within the set of clusters, as well as external cluster sinks, and possible ionization and recombination processes in the case that the simulation includes also charged clusters. As input, the rate constants of all the kinetic processes are required. For instance, quantum chemical thermodynamic data can be used in the ACDC framework by converting the cluster formation free energies into evaporation rates. Simulations on the formation of sulfuric acid-dimethylamine-ammonia clusters using quantum chemical data as input have produced thus far the best agreement with experimental results. Furthermore, the model can provide information, such as the clustering pathways, which cannot be obtained from measurements. In general, the ACDC model can be used to study
- the cluster concentrations, formation rates and growth pathways in the presence of different compounds
- Kupiainen-Määttä et al., In review for Chemical Physics Letters, 2014
- Bork et al., Atmospheric Chemistry and Physics, Vol 14, pp 12023-12030, 2014
- Almeida et al., Nature, Vol 502, pp 359–363, 2013
- Olenius et al., Faraday Discussions, Vol 165, pp 75–89, 2013
- Olenius et al., Journal of Chemical Physics, Vol 139, 084312, 2013
- Kupiainen et al., Atmospheric Chemistry and Physics, Vol 12, pp 3591-3599, 2012
- the general behavior of a cluster population in different conditions to test the performance of data analysis methods or more simplified clustering theories
- the effect of measurement methods, such as chemical ionization
Chemistry of Clusters
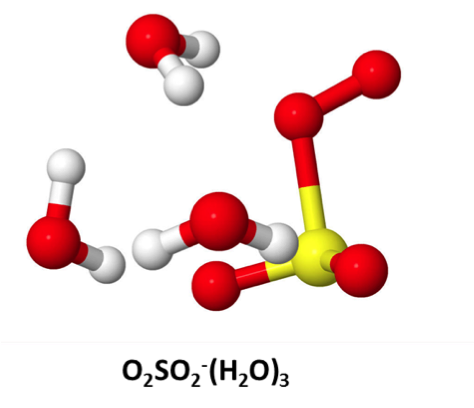
The local environment inside or on the surface of any aerosol is very different from the free gas phase. Hence, a number of chemical reactions may be facilitated by the presence of aerosols, leading to a changing composition of the gas phase or the aerosol itself. Until now, experimental studies of aerosol induced chemistry has been limited to large aerosol particles from primary emissions, e.g. dust or sea salt. However, we are interested in secondary particles, i.e. nanometer sized particles originating from gaseous phase. For these small particles experimental studies are difficult and rare, but they are small enough that the reactions may be studied using ab initio methods. We calculate the energy barriers, separating reactants and products, also known as the activation energy. By combining this knowledge with statistical physics the rate of the chemical reaction may be calculated as function of temperature. Hereby, the importance of the reaction may readily be determined at varying temperature, altitude and atmospheric composition, and used for input in the ACDC code or for explaining experimental or field data. Related recent publications from the group:
- Tsona et al. Atmospheric Chemistry and Physics, Vol. 15, pp, 495-503, DOI:10.5194/acp-15-495-2015, 2015.
- Bork et al. Physical Chemistry Chemical Physics, Vol. 16, pp, 24685-24690, DOI:10.1039/c4cp03828b, 2014.
- Tsona et al. Physical Chemistry Chemical Physics, Vol 16, pp 5987-5992, DOI: 10.1039/C3CP54715A, 2014.
- Bork et al.,Atmospheric Chemistry and Physics, Vol 13, pp 3695-3703, 2013.
Molecular Dynamics Simulations
Although ab initio methods applied with statistical physics provide excellent tools for predicting cluster formation, the huge number of possible molecular configurations of the clusters remains to be a challenge. Instead of high precision calculations on a few important configurations, another approach is to let the system evolve in time by numerically solving the equations of motion. Thereby, a realistic movie of the actual dynamics of a cluster of molecules is obtained. Such first-principles molecular dynamics simulations allow the detailed investigation of the dynamics of equilibrium clusters. Furthermore, also the likelihood of a given event, such as evaporation, fragmentation, oxidation or a "sticky" molecule-cluster collision, can be studied.
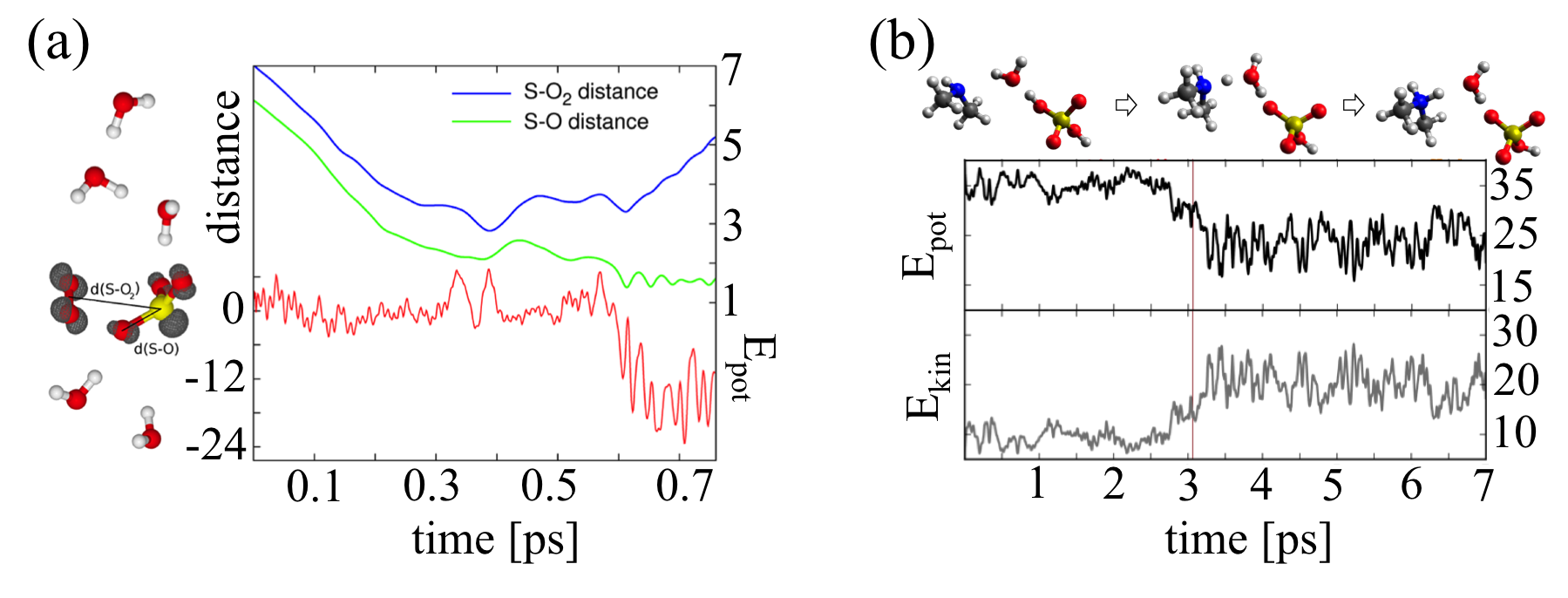
(a) Molecular dynamics simulation of a SO2 molecule collision with a O3 - (H2O)5 cluster. SO3 - is rapidly formed and the new O2 molecule is ejected from the cluster due to the high reaction entropy. (b) An example of a collision between sulfuric acid monohydrate and dimethylamine molecule. Here, the water molecule mediates the proton transfer reaction.
We are also using classical molecular dynamics simulations to study heterogeneous nucleation of ice, looking at kinetic and thermodynamic factors controlling nucleation at different conditions and in the presence of different surfaces. As ice nucleation is a rare stochastic process, a molecular-level understanding of the ability of aerosol particle surfaces to initiate ice nucleation is still lacking, even when this heterogeneous nucleation process is critical for formation of ice in clouds at temperatures above the homogeneous ice nucleation temperature, -37 °C. Surfaces with a good lattice match with hexagonal ice can be found to nucleate ice at MD time scales (in the order of 100 ns) in all-atom simulations of immersion mode nucleation.
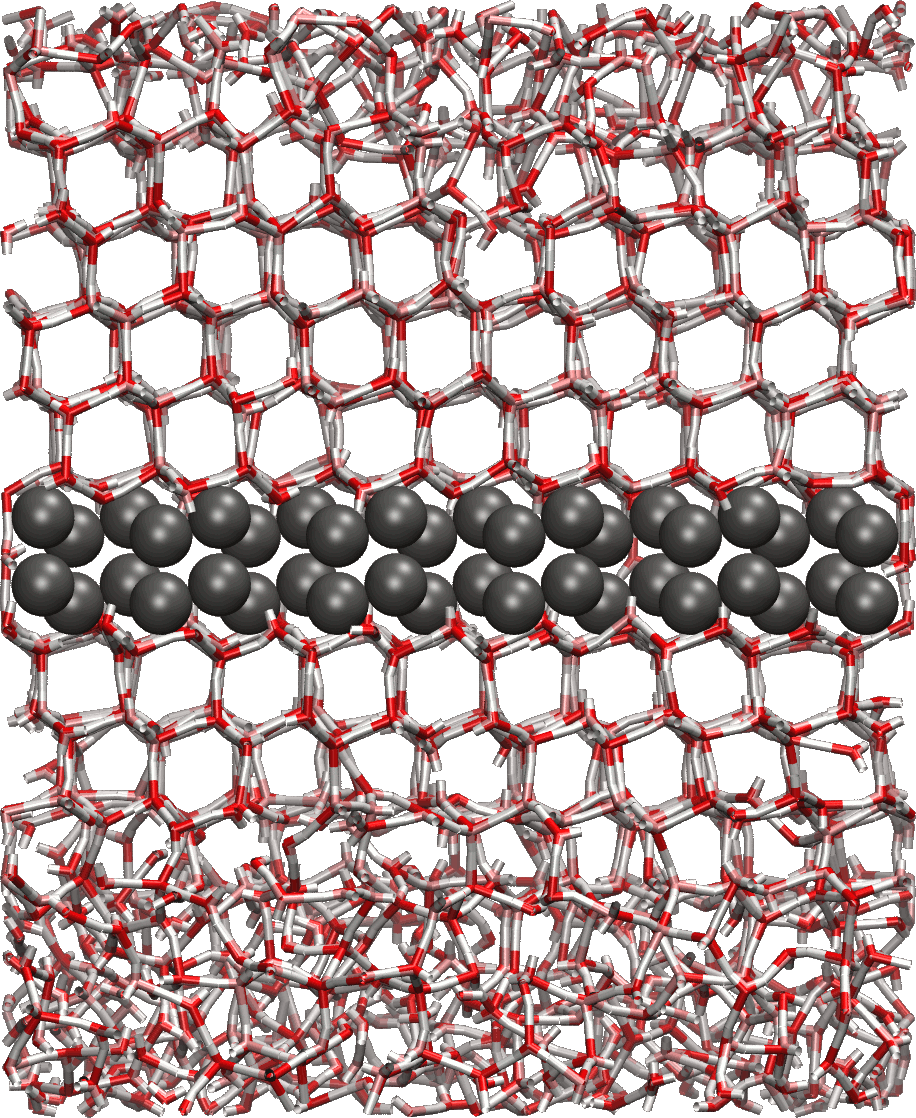
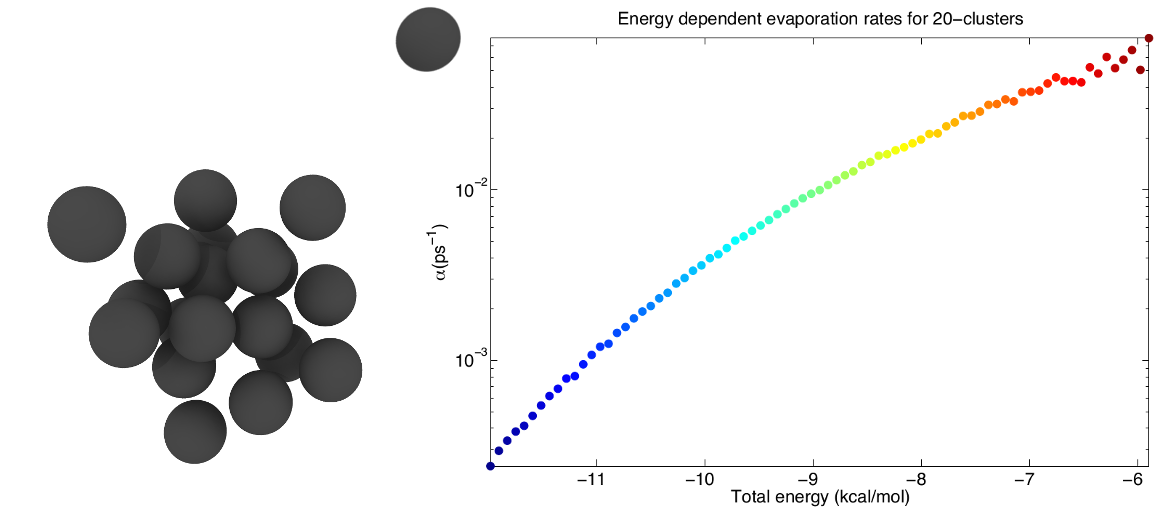
Furthermore classical MD is applied to demonstrate the incorrect description of small clusters of classical nucleation theory. One of the inherent assumptions of the nucleation models is the use of equilibrium thermodynamic methods in order to calculate the evaporation rates of different clusters. In our work the undemanding Lennard-Jones argon is used to study the nucleation of small non-equilibrium clusters, whose evaporation rates are highly dependent upon the total energy of the cluster.
Recent publications:
- Loukonen et al. Molecular Physics, Vol 112, pp. 1979-1986, DOI: 10.1080/00268976.2013.877167, 2014.
- Loukonen et al. Chemical Physics, Vol 428, pp. 164-174, DOI: 10.1016/j.chemphys.2013.11.014, 2014.
- Bork et al., J. Phys. Chem. A, 2013, 117 (15), pp 3143–3148
Classical Nucleation Theory and Related Approaches

Figure: An illustration of the formation free energy barrier to cluster growth.
Although the above mentioned methods are all state-of-the-art, the severe computational scaling of any ab initio method effectively prevents these methods from being applied to clusters larger than a few nanometers, containing at most 10-15 molecules. Entirely different from the explicit molecular modeling of ab initio approaches, classical nucleation theory (CNT) treats molecular clusters as spherical droplets consisting of bulk liquid. CNT is thus best applied to larger clusters more similar to bulk liquids. We have also experience on more advanced classical theories taking some of the micro-structure of the clusters into account. Quantum chemical data for the smallest clusters can be combined with CNT for larger clusters enabling the development of a typical cluster to be followed, starting from its initial clustering to the point where the cluster is large and thermally stable. Even thermodynamic theories like CNT are computationally too expensive for use in larger scale atmospheric models, and results have to be parameterized to reduce computational cost. Related recent publications from the group:
- Vehkamäki et al., J. Chem. Phys., 136, 094107, 2012
- Vehkamäki and Riipinen, Chemical Society Reviews, Vol 41 (15), pp 5160-5173, 2012